

Paper translation of “Predicting locations of cryptic pockets from single protein structures using the Pocket Miner graph neural network”
结果
很少有研究系统地评估分子动力学(MD)模拟是否概括了已知的神秘口袋。在之前的工作中,MD 模拟捕获了 TEM β-内酰胺酶 22 、Interleukin-2 23 和其他几种蛋白质药物靶标 13,24 中已知隐秘位点的开放。 MD 模拟还发现了新的神秘口袋,这些口袋在硫醇标记实验中得到了实验证实 5,25 。然而,这些研究集中于一小部分蛋白质,或者没有评估口袋形成是否局限于已知的配体结合位点。鉴于最近人们对使用 MD 模拟来发现神秘口袋的兴趣,有必要对 MD 模拟重现已知神秘口袋的效果进行系统评估。这样的研究也可以生成数据来训练机器学习模型来识别神秘口袋形成的位置。
因此,我们对已知从 apo(或无配体)起始结构形成隐秘口袋的 16 种蛋白质进行了无偏自适应采样 MD 模拟。其中 11 对具有配体结合残基,它们在 apo 结构中比在holo结构中更靠近(即,它们需要打开运动才能形成口袋)。代表了多种不同类型的运动,包括二级结构变化的三种情况。对于每种蛋白质,我们使用特定性状波动放大 (FAST) 算法 26 运行 2 μs 的自适应采样模拟。具体来说,从apo结构发起了10次并行模拟,然后构建了马尔可夫状态模型(MSM)的构象集合和优先结构用于下一轮模拟(即,优先考虑具有大口袋的状态)。此过程重复四次以生成5个“swarms”模拟,每个模拟包含400ns的聚合模拟(10个模拟长度为40ns)。
令我们惊讶的是,我们发现绝大多数神秘口袋仅在 40 纳秒的 10 次并行模拟中就打开了(图 2)。如果模拟结构的袋体积达到或超过holo结构袋体积,则认为袋是开放的(参见方法)。其中一种蛋白质的隐秘体积在 apo 中比在全息中更大,因此在进一步分析中被排除。其余 15 个蛋白质中有 13 个在 40 纳秒的前 10 次模拟中打开。另一项是伸长因子 TU,在额外几轮自适应采样(五轮 10 次并行 40 ns 模拟)后打开。即使在 5 轮自适应采样模拟之后,只有一种蛋白质 Niemann-Pick C2 蛋白质仍然保持关闭状态。我们假设该蛋白质没有观察到隐秘袋开口,因为结合在隐秘袋中的配体是高度疏水性的 29 。令人鼓舞的是,我们还发现模拟中形成的隐秘袋大多位于已知的配体结合隐秘位点,其中最大的袋体积变化发生在配体结合位点(补充图2,补充表6)。总而言之,我们观察到的口袋快速打开表明,对于大多数较小的蛋白质来说,适量的模拟数据可能足以发现神秘的口袋。此外,这一发现表明,经过训练以在小模拟时间窗口(即 40 ns)内预测隐秘袋形成的机器学习模型也能够识别无配体实验结构中的隐秘位点。
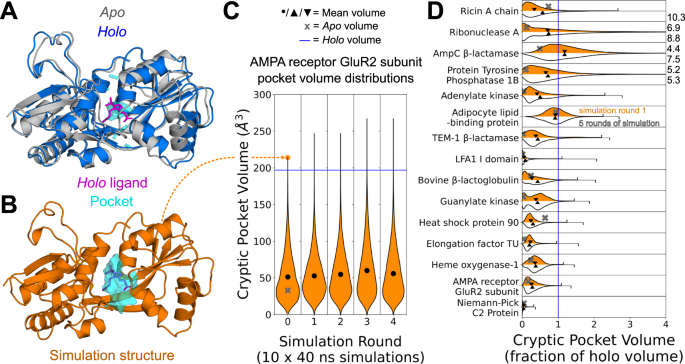
图 2:在从封闭的 apo 结构开始的模拟中,隐秘袋迅速打开。
AMPA受体蛋白GluR2亚基在Apo(灰色,PDB:1MYO)和Holo(蓝色,PDB:1N0T)构象中的结构叠加表明,环和螺旋必须移动才能形成隐蔽的口袋。B来自MD模拟的结构从Closed的apo构象开始,在结合位点有一个口袋,其体积超过holo结合位点口袋(橙色)。在模拟AMPA受体的GluR2亚基时,C. MSM加权小提琴图表明配体结合位点超过了holo中看到的体积,仅10个平行模拟,每个模拟40ns。通过将每个Ligsite网格点分配给最近的单个残基并将分配给所有隐蔽口袋残基的体积相加来计算隐蔽口袋体积。灰色”X“表示apo结合位点隐蔽口袋体积,蓝色表示holo体积,黑点表示每一轮模拟所有帧的MSM加权平均体积。D. MSM加权的15种不同蛋白质模拟的隐蔽口袋体积分布表明,即使10次短模拟通常也足以达到holo结构的隐蔽口袋体积。作为holo的一部分口袋体积分布10次短模拟的体积以橙色显示,而5轮自适应采样分布以白色显示。灰色“X”表示apo隐袋体积作为holo的一部分,向下和向上的三角形分别表示10次模拟(1轮)或5轮自适应采样后的MSM加权平均体积。 模拟中达到的最大神秘口袋体积作为holo的一部分用刻度线显示,如果它们位于图的范围之外,则在图的右侧以数字形式给出。源数据作为源数据文件提供。
图神经网络准确预测模拟中口袋的时间演化
给定一个起始结构,我们推断,在 MD 模拟过程中,人们可以预测该结构中的神秘口袋将在哪里形成。尽管 MD 模拟是随机的,并且从同一结构启动的两个模拟不会遵循完全相同的时间演化,但我们推断足够长的模拟将采样类似的口袋打开事件。例如,一个与邻近残基之间相互作用少且松散排列的残基可能会在多次模拟中移动,形成一个持续存在的口袋。相反,与邻居紧密相互作用的残基更有可能在独立模拟中保持锁定在其初始位置。
为了确认我们的直觉,我们评估了已知的隐藏口袋在独立模拟中口袋打开的一致性。我们使用LIGSITE算法计算了每个模拟中访问的结构的口袋体积(每个40纳秒模拟2000个结构),并将口袋体积分配给附近的残基。具体而言,我们确定了与每个残基相距不超过5埃的LIGSITE口袋格点的数量。然后,我们将残基二值化为参与口袋开启事件和未参与形成口袋的残基。如果在模拟中的任何时刻,由LIGSITE算法确定的附近口袋体积相对于起始结构中的分配口袋体积增加了40埃以上,那么将残基视为正例(请参阅方法)。我们发现,从相同的起始结构启动的独立40纳秒模拟产生了类似的标签,绝大多数残基要么总是开放,要么总是关闭(补充图3)。当我们对像TEM 1 β-内酰胺酶这样已知存在隐藏口袋开启的蛋白质进行视觉检查时,我们观察到在多次模拟中相似的活性位点中存在开启现象(附图2)。
鉴于独立模拟会收敛到相似的标签,我们着手开发一种算法来预测模拟中神秘口袋打开事件的地点。具体来说,我们将已知神秘口袋开口的模拟数据与 SARS-CoV-2 蛋白的附加自适应采样模拟相结合 12 。该数据集包括 37 种蛋白质和 2400 个长度至少为 40 ns 的独立 MD 模拟。我们根据残基是否在接下来的 40 ns 模拟中的任何点参与神秘口袋,为每个 40 ns 窗口的起始结构中的每个残基生成标签(图 3A)。该数据集总共包含 941,650 个独特的示例。然后,我们使用这些数据来训练基于几何矢量感知图神经网络 (GVP-GNN) 31 以及 3D 卷积神经网络 (3D-CNN) 32
图 3:图神经网络在新模拟中准确预测了口袋形成的位置。
A:我们使用 MD 模拟通过跟踪神秘口袋(以红色显示)的形成位置来生成训练标签。如果在模拟的任何时刻某残基附近形成了一个隐蔽口袋,则将该残基标记为正样本(用青色显示)。为了说明,我们展示了TEM β-内酰胺酶蛋白,它在单个MD模拟中形成了两个分离的隐蔽口袋(即角口袋和Ω环口袋)。每个开放事件都会将附近的残基标记为正样本,因此该窗口的训练标签反映了两个开放事件,两个口袋周围的残基都标记为正例。然后可以将起始结构输入到图神经网络中来预测隐蔽口袋将形成的位置。
B:在5个不同的折叠中,精确度-召回曲线表明,使用模拟的起始结构和来自模拟中间体的标签训练的图神经网络可以预测哪些残基将在模拟中形成隐蔽口袋(平均PR-AUC为0.44)。阴影区域表示在不同折叠中性能的变化,阴影区域的顶部描绘了具有最高PR-AUC的曲线,底部描绘了具有最低PR-AUC的曲线。GVP-GNN指的是基于几何向量感知器的图神经网络;3D-CNN指的是3D卷积神经网络。
C:在5个不同的折叠中,ROC曲线展示了稳健的性能(平均ROC-AUC为0.83)。阴影表示折叠间的变化(5个折叠中性能从最差到最好)。源数据提供在一个源数据文件中。

我们发现,GVP-GNN 和 3D-CNN 都能够根据起始结构在模拟中准确分类给定的残基是否会形成神秘口袋。我们将 37 个蛋白质模拟数据集按蛋白质分为 5 折,并使用 5 折交叉验证来衡量两种架构的性能。对于每次分割,我们分别评估了模型超参数(例如,退出率)和训练设置(例如,类平衡方案)中的选择如何影响 1 折(3 折用于训练)的性能。然后使用保留的测试折(补充表 1)评估在特定分割的每个验证折叠(补充表 2-4)上表现最佳的 GVP-GNN 和 3D-CNN。在 5 个分割中,最佳 GVP-GNN 模型的平均测试 PR-AUC 为 0.44 ± 0.12(平均 ROC-AUC:0.83 ± 0.04;图 4B-C)。 3D-CNN 的表现类似(PR-AUC:0.41 ± 0.05;ROC-AUC:0.79 ± 0.02)。在高召回率水平 (0.6-0.8) 下,GVP-GNN 做出的误报预测较少,正如精确率-召回率曲线右侧的更高精确率所证明的那样(图 4B)。因此,我们决定继续使用 GVP-GNN 架构来完成进一步的预测任务。
总体而言,这些发现表明,当给定蛋白质的天然折叠状态结构时,无需计算中间状态(例如,通过 MD 模拟)即可识别神秘口袋形成的位点。
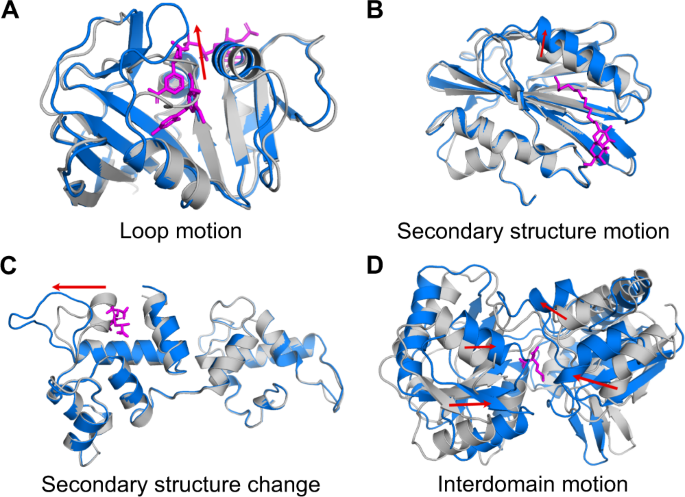
Apo 结构以灰色显示,holo结构以蓝色显示,配体以洋红色显示。红色箭头突出显示了 apo 和 Holo 之间发生的主要构象变化。
(A) 隐性口袋可能是由于循环运动而产生的,就像二氢叶酸还原酶中所见的那样(apo PDB:2W9T 74 ,holo PDB:2W9S 74 );
(B) 二级结构元件运动,例如脂蛋白 LpqN 中所见的运动(apo PDB:6E5D 75 ,holo PDB:6E5F 75 );
(C) 二级结构变化,类似于钙结合蛋白和整联蛋白结合蛋白 1 中观察到的变化(apo PDB:1Y1A 链 A 76 ,holo PDB:1Y1A 链 B);
(D) 或胭脂碱结合周质蛋白中的域间运动(apo PDB:4P0I 77 ,holo PDB:5OTA 78 )。我们将 A-C 中所示的例子称为前向口袋,因为全息结构中与配体相邻的残基彼此之间的距离比在 apo 结构中的距离更远。我们将 D 中的例子称为反向口袋,因为 Holo 中与配体相邻的残基之间的距离比 apo 中的更近。
隐秘口袋数据集揭示了向前和向后的运动
为了评估我们的模型是否能够从实验证实的配体结合隐蔽口袋中检测到隐蔽口袋形成的位置,我们首先整理了一个隐蔽口袋的数据集。Cimermancic等人之前已经识别出93个含有隐蔽口袋的apo-holo蛋白质结构对,这里将其称为CryptoSite集合。虽然有用,但CryptoSite集合中包含的蛋白质相对较少,其中需要大的构象变化才能形成口袋。此外,自生成CryptoSite集合所用的PDB版本以来,PDB的大小大约翻了一番,Sun等人观察到CryptoSite集合中的一些蛋白质有额外的apo结构,其中口袋是打开的。我们推断,整理一个新的隐蔽口袋数据集将改善我们对隐蔽口袋的理解,并为我们的模型提供一个测试集,该测试集对隐蔽口袋结构和运动的大而多样的空间具有增加的覆盖。因此,我们筛选了蛋白质数据银行(PDB),以识别出38个apo-holo蛋白质结构对,这些蛋白质结构对中,包含了39个隐蔽口袋,这些口袋在apo结构和holo结构之间具有较大的根均方偏差。(蛋白质的apo状态(未结合配体)和holo状态(已结合配体)之间的结构变化较大。这种变化可以通过计算apo结构和holo结构之间的根均方偏差来量化,而这些口袋具有较大的根均方偏差表示它们在结构上发生了显著的变化。)
由此产生的神秘口袋集合称为 PocketMiner 数据集,其中包括由多种构象变化形成的口袋。有趣的是,我们注意到许多口袋是通过关闭动作而不是典型的口袋打开动作形成的。对于这些“反向”口袋,结构元件开始时相距很远,因此 apo 结构中几乎没有或没有口袋,但聚集在一起形成盖子或壁,形成一个空腔,配体可以在全息中结合(图 1)。 4D)。大多数型腔主要由单一类型的运动形成,尽管在某些情况下多种类型的运动也有显着的作用。在前袋和后袋中,我们观察到四种常见的结构重新排列。首先,环可以分开,为进入的配体创造空间,或者夹紧以在配体上形成壁或盖(图4A)。其次,二级结构元件可以通过平移和/或旋转来移动(图4B)。第三,二级结构元素可以变成环或形成环(图4C)。第四,域间运动可以为配体结合创造空间(图4D)。因此,我们神秘的口袋数据集捕获了一组不同的构象重排,并代表了评估机器学习模型的具有挑战性的基准。
PocketMiner 从无配体的晶体结构中准确预测配体结合的隐秘口袋
考虑到GVP-GNN准确地预测了模拟中隐蔽口袋的形成位置,我们想知道这种网络架构是否可以用于预测实验结构中隐蔽口袋形成的位置。如果已知的隐蔽口袋在模拟中迅速形成,那么一个学会预测口袋形成位置的GVP-GNN可能合理地识别实验结构中的配体结合隐蔽位点。(注:模拟生成的数据和真实实验获得的数据)
因此,我们从之前的训练集中获取了所有蛋白质,并根据 LIGSITE 口袋体积和 fpocket 成药性分数的局部变化生成训练标签 35 (参见方法)。我们决定使用 fpocket 标记方案,因为成药性分数不仅考虑口袋的几何形状,还考虑口袋的化学环境。我们仅使用 LIGSITE 衍生标签、仅使用 fpocket 衍生标签以及两种标签的组合来训练 GVP-GNN(即,使用一种标签方案对网络进行多个时期的训练,然后切换到从其他标签学习多个时期)更多纪元)。尽管之前的研究发现 LIGSITE 和 fpocket 很难正确排列哪些口袋结合配体 36 ,但我们的标签没有考虑单个结构中口袋之间的排名。相反,我们的标签是基于 MD 模拟过程中 LIGSITE 口袋体积的变化或残留物附近的最大 fpocket 成药性分数。然后,我们评估使用这些标签训练的模型是否可以区分配体结合隐性位点的残基和不形成隐性口袋的残基。
因此,我们从先前的训练集中选取了所有的蛋白质,并基于LIGSITE口袋体积的局部变化和fpocket成药性分数生成了训练标签(见方法)。我们决定使用fpocket标记方案,因为药物成药性分数不仅考虑了口袋的几何形状,还考虑了口袋的化学环境。我们使用了仅基于LIGSITE导出的标签进行训练,仅基于fpocket导出的标签进行训练,以及使用两种标签的组合进行训练(即,网络首先使用一种标记方案进行数个epochs的训练,然后切换到另一种标记方案进行数个epochs的训练)。虽然先前的研究发现LIGSITE和fpocket可能会在正确排列结合配体的口袋时遇到困难,但我们的标签并未考虑单个结构中不同口袋之间的排名。相反,我们的标签是基于MD模拟过程中某个残基附近的LIGSITE口袋体积变化或最大fpocket药物成药性分数的变化。然后,我们评估了使用这些标签进行训练的模型是否能够区分配体结合隐蔽位点上的残基与不形成隐蔽口袋的残基。
为了评估我们的模型是否能够区分不形成隐蔽口袋的位点,我们整理了一个负样本数据集,其中包括刚性蛋白质和其他进行了大量药物筛选的蛋白质。在先前的工作中,负样本被定义为任何不参与配体结合隐蔽口袋的残基21。然而,许多这些位点可能具有形成配体结合隐蔽口袋的潜力。为了增加对我们负样本的信心,我们仅使用了已知极其刚性或极其稳定的蛋白质的残基,或者来自已经进行了大量药物筛选的蛋白质的残基。我们的超刚性蛋白质数据集包括三个设计的微蛋白质和一些具有异常刚性折叠或类晶体属性的蛋白质(见方法)。我们对这些蛋白质进行了MD模拟以验证其不形成隐蔽口袋,并从模型评估数据集中移除了任何附近有小口袋的残基(见方法)。为了补充这些用于模型评估的相对较少的负样本数量,我们还识别了曾进行大量药物筛选的蛋白质,并提取了配体未结合的残基(补充图5)。我们的推理是,配体未结合的位点极不可能形成隐蔽口袋,因为这些蛋白质已经与大量配体共结晶或浸泡。为了对我们的负标签建立额外的信心,我们还对这些蛋白质进行了模拟,并移除了在模拟中形成隐蔽口袋的任何残基(补充图6)。因此,我们汇集了一系列具有实验和模拟证据支持的负样本。
我们发现我们的最终模型(称为 PocketMiner)在区分形成神秘口袋的残基和不形成神秘口袋的残基方面取得了非常好的性能(ROC AUC:0.87)。 PocketMiner 使用源自 LIGSITE 的标签进行了 20 个 epoch 的训练,并使用源自 fpocket 成药性分数的标签进行了 1 个 epoch 的细化。为了测试 PocketMiner,我们对总共 24 个形成配体结合隐秘口袋的 apo 结构、4 个超刚性蛋白和 7 个作为广泛配体筛选对象的蛋白进行了预测。测试集中的所有蛋白质与训练集中的蛋白质的序列同一性均低于 55%(补充图 7、补充表 5)。在我们的测试集中,总共有 563 个形成隐秘口袋的残基和 1283 个不形成隐秘口袋的残基。在形成隐秘袋的apo结构中,我们的模型预测在实验隐秘位点形成隐秘袋的可能性很高(图5A,补充图7)。我们在几类神秘口袋open中实现了稳健的性能(图5C,补充图7,补充表9)。相反,我们的模型预测所有超刚性蛋白质形成隐秘袋的可能性较低,并正确地将低预测分配给不太可能由具有广泛配体结合的蛋白质形成隐秘袋的区域(图5B,补充图8)。
当我们将模型的性能与 CryptoSite 进行比较时,我们发现这两种方法的性能非常相似,PocketMiner 提供了一点优势(ROC-AUC:PocketMiner 为 0.87,而 CryptoSite 为 0.85)。 特别是,我们发现 PocketMiner 在刚性蛋白质和不结合配体的位点中预测的误报较少,这表明 PocketMiner 可能是一种更有用的筛选工具(补充表 8)。此外,我们注意到,虽然 CryptoSite 必须运行通常需要几个小时才能生成预测的模拟,但我们的模型在一秒钟内即可做出预测(预测时间提高了 1000 倍以上)。
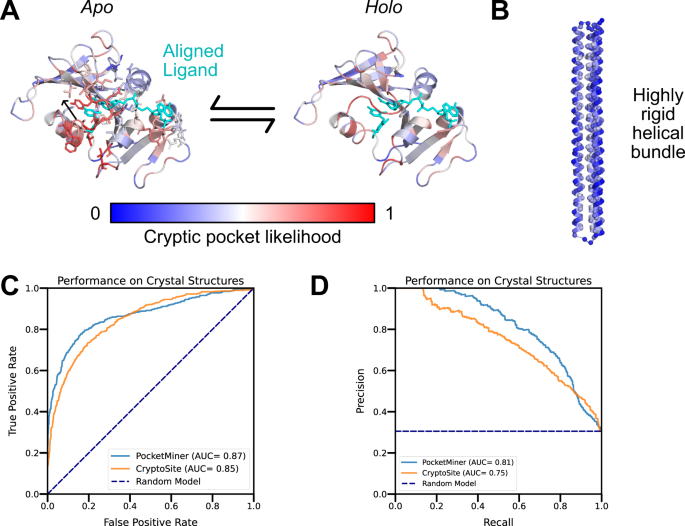
图 5:PocketMiner 图神经网络准确检测实验结构中神秘口袋形成的位置
A PocketMiner预测在配体结合位点存在隐蔽口袋形成的可能性很高。来自对齐的holo结构的配体(青色)显示在apo结构上(配体(药物分子或配体)在holo结构中被定位,然后将其位置与apo结构进行对齐,即将药物分子在holo结构中的位置映射到apo结构上),以突出配体与必须移动以创建holo结合位点的环之间的立体阻碍。尽管PocketMiner仅使用apo结构生成预测(蓝色表示隐蔽口袋形成概率较低,而红色表示概率较高),但预测的标签也显示在holo结构上,以突显那些高预测标签在配体结合位点附近的聚集。
B PocketMiner正确地预测了高度刚性螺旋捆绑体(PDB:4TQL79)在模拟中没有形成大的隐蔽口袋的概率较低。
C 残基级别隐蔽位点检测的接收器操作曲线显示,尽管运行速度快了1000倍以上,PocketMiner的性能优于CryptoSite。
D 精确度-召回曲线突显出在高召回率水平(0.6–0.8)下,PocketMiner预测的假阳性更少。源数据提供在一个源数据文件中。
PocketMiner 预测人类蛋白质组中存在数千个神秘口袋
鉴于其能够准确、快速地预测隐秘袋的位置,PocketMiner 可用于在大量蛋白质结构中搜索隐秘袋。得益于结构生物学数十年的努力以及AlphaFold 2 19 实现的高精度蛋白质结构预测,标准人类基因组中所有基因的蛋白质结构都被提出。我们假设 PocketMiner 可以帮助发现针对人类蛋白质设计药物的新机会,这些蛋白质在其天然折叠结构中缺乏清晰的药物结合口袋。即使在已经存在明确结合袋的情况下,PocketMiner 也可以识别蛋白质其他区域中的隐秘袋,这些区域可以对蛋白质功能施加变构控制,或用于设计具有更好特异性的药物。
为了在人类蛋白质组中识别隐蔽口袋,我们将PocketMiner应用于超过10,000个人类基因。对于每个基因,我们优先选择使用高分辨率的实验确定结构,但在没有可用的实验结构时,我们使用AlphaFold预测的结构作为替代方案。最后,我们丢弃了具有长段预测无序的结构(见方法)。我们将结构分成三类:具有基态口袋的蛋白质、仅具有隐蔽口袋的蛋白质,以及两者都缺乏的蛋白质。根据计算其最大LIGSITE口袋体积和其最高PocketMiner预测,对每个结构进行分组(见方法)。
我们发现,尽管在它们的基态结构中缺乏口袋,但有几千种蛋白质被预测具有隐蔽口袋(在我们的数据集中占29.4%,如图6A所示),将可能形成口袋的单结构域蛋白质的比例扩大到超过80%。根据我们的分析,一小部分单结构域蛋白质既缺乏基态口袋又缺乏隐蔽口袋(占18.5%)。预测到的大量隐蔽口袋表明,即使在其原生、折叠结构中没有明显的小分子结合位点,对蛋白质进行药物筛选可能也是有成果的。理想情况下,我们会在一些具有治疗兴趣的蛋白质组中使用PocketMiner,确定哪些蛋白质可能具有隐蔽口袋,然后模拟该蛋白质的结构动态,以识别其结构集合中采用的可药用结构。PocketMiner的残基水平口袋网站预测可以使用自适应或增强的采样方法,针对 包含隐藏口袋的蛋白质区域 进行预测。
图 6:在人类蛋白质组中应用 PocketMiner 揭示了数千个神秘的口袋。
A. 虽然将近一半的蛋白质在其原生、折叠状态中缺乏口袋,但其中大多数蛋白质被预测具有隐蔽口袋。饼图显示了 含有口袋的蛋白质以小麦色显示( Pie chart shows the number of human proteins containing pockets in their native structures in wheat; );在其原生结构中缺乏口袋但可能形成隐蔽口袋的蛋白质以红色显示;在其原生结构中既缺乏基态口袋又缺乏隐蔽口袋的蛋白质以灰色显示。
B. 示意图显示了与癌症相关的Jak/Stat信号通路中涉及的蛋白质。
C. PIM2激酶的晶体结构包含一个正交配体结合位点,但没有显示任何变构口袋。
D. PocketMiner预测在一个变构位点上存在一个隐蔽口袋。
E. 模拟重现了PocketMiner预测的一个隐蔽口袋。在模拟中,一个环被拉开以显示由PocketMiner确定的一个隐蔽口袋(以青色显示)。具有一个隐蔽口袋的模拟PIM2结构(以橙色显示)与apo PIM2结构(以灰色显示)重叠。
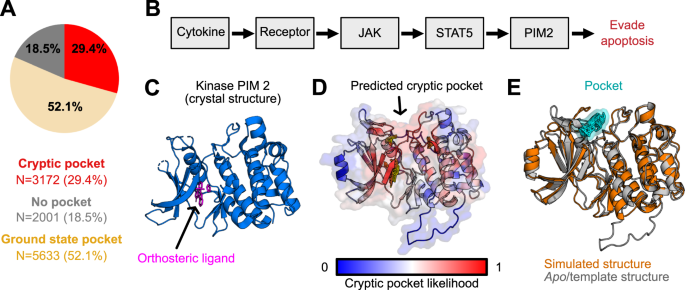
为了展示PocketMiner的实用性,我们使用它来帮助识别涉及到多种癌症的Jak/Stat通路中的蛋白质中的隐蔽口袋37。首先,我们将PocketMiner应用于KEGG通路中称为“癌症途径”的所有蛋白质(KEGG ID:05200)。我们确定了PIM2具有高隐蔽口袋预测。PIM2是一个已知的丝氨酸/苏氨酸激酶原癌基因,其已知的正交配体结合位点已成为药物筛选的靶点39。PocketMiner预测在活性位点“上方”存在一个隐蔽口袋,可能可用于设计更具特异性的化合物(图6D)。我们对PIM2进行了模拟,并发现隐蔽口袋确实在预测的区域形成(图6E)。重要的是,这个口袋之前从未被小分子靶向过,但从模拟中得出的结构可能有助于引导药物开发活动。在KEGG通路中,还有其他一些蛋白质,比如WNT2,在其基态结构中缺乏口袋,但有高隐蔽口袋预测。PocketMiner预测WNT2具有隐蔽口袋,并且我们对该蛋白质的模拟捕获了在预测位点形成一个大的口袋(补充图9)。考虑到WNT2缺乏任何实验结构,这表明将PocketMiner应用于AlphaFold结构是扩展可药用蛋白质集合的可行策略。
方法
分子动力学模拟
系统准备
被选定用于模拟的蛋白链的结构从RCSB PDB下载。这些结构被加载到PyMOL中,只有蛋白质链被保存以生成模拟输入结构(在将结构加载到PyMOL软件中后,只有蛋白质链被保留下来,而其他非蛋白质的部分(如水分子、离子等)被移除,从而生成了用于进行模拟的输入结构。这是因为在许多模拟程序中,通常只考虑蛋白质的结构,而将其他非蛋白质的组分排除在外可以简化模拟系统,并且有助于减少计算的复杂性。)。未解决的内部环被使用SWISS-MODEL建模(使用蛋白质的氨基酸序列建立预测的蛋白质三维结构),使用可用的晶体结构作为模板,并使用相同RCSB PDB条目的FASTA序列作为目标序列。未解决的末端区域未被建模。
注:(模型的建立和补全)
- 使用可用的晶体结构作为模板:这意味着在建立模型时,使用已知的晶体结构作为参考,以便构建一个与已知结构相似的模型。这有助于确保新模型的准确性和可靠性。
- 使用相同RCSB PDB条目的FASTA序列作为目标序列:在建立模型时,将与晶体结构相对应的FASTA序列作为目标序列,以便在模型中保持与实际蛋白质序列的一致性。
- 未解决的末端区域未被建模:这指的是在模型构建过程中,由于数据不完整或不足,无法建立的末端区域未被包括在内。这些未解决的区域可能会导致模型的不完整或不准确,因此在模拟中可能会忽略这些部分。
本研究中的所有模拟均使用GROMACS(Gromacs 2020.1)进行准备。蛋白拓扑结构(拓扑结构是指描述分子内部原子之间连接方式以及它们之间相互作用的网络。)使用Amber03 进行准备。(使用 Amber 而不是 GROMACS 进行蛋白质拓扑结构的准备是因为 Amber 是一种力场,它定义了模拟中原子之间的相互作用参数,如键长、键角、二面角等。在分子动力学模拟中,选择适合的力场是非常重要的,因为它会直接影响模拟结果的准确性和可靠性。虽然 GROMACS 也提供了一些内置的力场参数,但 Amber 力场被认为是较为经典和广泛应用的力场之一,特别适用于蛋白质和核酸的模拟。因此,研究人员经常选择使用 Amber 来准备蛋白质的拓扑结构,以确保模拟结果与实验数据的一致性,并且在一些研究中,特定的力场可能更适合描述所研究的体系。)在生产分子动力学过程中使用虚拟位点45,以允许每步4 fs的时间步长。蛋白质被溶解在一个TIP3P水46的菱形十二面体盒子中,蛋白质与盒子边缘之间的最小距离为1 nm。Na+ 和 Cl- 离子被添加以中和系统的净电荷,并达到0.1 mol/l的盐浓度。
使用初始步长为 0.01nm 的最速下降算法最小化蛋白质的势能。最小化一直持续到最大力降至 100kJ/(mol*nm) 以下或持续 500 步。
蛋白质以 2 fs 时间步长平衡 0.1 ns。所有键均使用 LINCS 算法 47 进行约束。邻居列表使用 Verlet 截止方案和 1.1 nm 截止半径。库仑和范德华相互作用使用 0.9 nm 的截止值。使用粒子网格 Ewald 方法 48 处理长程相互作用,傅立叶间距为 0.12 nm。使用速度重新调整恒温器 49 ,温度设置为 300 K。
分子动力学模拟
本研究中使用了GROMACS(Gromacs 2020.1和Gromacs 2021.2)对所有蛋白质进行了模拟。采用了4 fs的时间步长。利用leap-frog算法来积分牛顿运动方程。所有涉及氢的共价键都使用LINCS算法进行约束47。库仑和范德华相互作用的截断距离为0.9 nm。采用粒子网格Ewald方法来处理长程相互作用48,傅立叶间隔为0.12 nm。采用速度重标定恒温器49来保持系统温度在310 K,采用Parinello-Rahman压力控制器50来维持系统在1 bar的压力下。
自适应采样协议
采用了自适应抽样模拟,使用了FAST算法26。FAST算法接受一个起始结构和一个排序参数,并使用多轮并行无偏分子动力学模拟来搜索最大化或最小化排序参数的构象。FAST的第一轮包括从单一结构开始的n个并行轨迹。在每一轮FAST结束时,迄今为止所有FAST轨迹的模拟轨迹帧通过RMSD进行聚类,然后根据排序参数对聚类中心进行排名。然后,选择排名前n的聚类中心用于开始下一轮FAST的n个并行轨迹。
注:
FAST算法指的是快速采样技术(Fast Adaptive Sampling Technique),它是一种用于蛋白质构象搜索的自适应采样方法。FAST算法使用多轮并行的无偏分子动力学模拟,以搜索在给定排序参数下最优的构象。通过迭代地从前一轮模拟的结果中提取信息并引导下一轮模拟,FAST能够快速地在构象空间中搜索到感兴趣的结构。该算法常用于寻找特定性质或结构的蛋白质构象,如蛋白质的折叠状态、配体结合构象等。
本研究中使用了每个蛋白质的平衡晶体结构作为FAST的起始结构。由于我们从模拟中生成准确的训练标签的能力取决于我们的模拟能否对隐蔽口袋的开放进行采样,而我们需要一个不依赖于口袋位置先验知识的排序参数,因此我们使用总的LIGSITE30口袋体积作为我们的排序参数。我们使用k-centers算法进行聚类,并使用所有主链α-碳之间的RMSD作为距离度量。对于每个模拟,我们确定了一个聚类半径,以产生100至300个聚类,并将相同的聚类半径用于额外的自适应采样轮次。我们进行了3、5或7轮自适应采样,具体取决于模拟。有关本研究中进行的所有模拟的完整列表,请参阅补充表4。本研究还使用了来自多个来源的预先存在的FAST模拟数据,其中并非所有数据都使用相同的FAST参数5,12。
Network architectures 网络架构
基于几何向量感知器的图神经网络(GVP-GNN)
PocketMiner 是一种等变图神经网络。这种基于几何向量感知器的图神经网络(GVP-GNN)最初设计用于学习蛋白质级表征。在我们的工作中,我们将该模型调整为学习残基级表征,从而推断出每个残基参与隐秘口袋的形成的概率。
GVP-GNN 通过相邻残基之间的信息交换来学习残基化学和拓扑环境的表示。具体来说,网络的输入包括 描述残基属性的节点特征和描述残基之间关系的边缘特征。
节点特征
- 标量特征 {sin, cos} ◦ {φ, ψ, ω},其中 φ, ψ, ω 是根据 C i−1 , N, C αi , C, 计算的二面角,和 N i+1 。
- Cβi - Cαi 推测方向上的单位向量。计算方法是假设四面体几何形状并进行归一化处理。
- 正向和反向单位向量。
- 氨基酸同一性的一次性表示。
边特征
是针对感兴趣残基的最近 30 个邻居计算的,包括以下内容:
- Cα − Cα 方向的单位向量。
- 用高斯径向基函数对距离 | |Cαj - Cαi | |2 进行编码。
- j - i 的正弦编码,代表沿主干线的距离。
网络如何在相邻残基之间交换信息以学习残基表示法
在此,我们将对网络如何在相邻残基之间交换信息以学习残基表示法进行说明性描述。有关精确的数学描述,请读者参阅原著。在每个图传播步骤中,残基 i 的表征都会根据其 30 个最近邻居的属性以及残基 i 与每个邻居之间的空间和序列关系进行更新。邻居的节点特征和残差 i 与邻居之间的边缘特征被拼接起来,因此对于单个残差 i,网络会收到 30 个向量。网络通过 “几何向量感知器”(见原论文)对这些向量进行转换,然后取 30 个输出向量的平均值,并根据残基 i 的节点特征将其添加到残基 i 的自我表示中,从而得到残基 i 的最新表示。残基 i(以及所有其他残基)会经历 n 次更新过程。迭代更新的结果是,残基 i 的表示受其邻居的邻居(以此类推)的隐性影响,因为残基 i 的表示是根据其邻居的表示计算出来的,而邻居的表示又受其邻居的影响。最终,每个残基都会得到自己的表征,该表征会考虑到整个蛋白质的拓扑结构和化学环境。
超参数
我们使用上述架构和以下超参数训练了 GVP-GNN 模型:
- 结构特征节点维度: 8 个向量维度,50 个标量维度。
- 结构特征边缘维度:1 个向量维度,32 个标量维度。
- GVP隐藏维度:16个向量维度,100个标量维度。
- 编码层:4。
- 图传播的邻居列表:30。
- Dropout rate: 0.1
3D 卷积神经网络 (3D-CNN)
(略)
GVP-GNN和3D-CNN模型训练和评估(任务1)
我们训练了两种不同的架构,目的是使用起始结构预测模拟中的口袋体积变化(任务 1)。
数据标签化
我们使用 MD 模拟中的蛋白质结构进行训练,每个残基都分配了一个标签,具体取决于在接下来的 40 ns 模拟中该残基附近是否形成了口袋。具体来说,我们使用 LIGSITE 口袋检测算法计算口袋,将口袋映射到残基,并确定每个残基在起始结构中分配的口袋体积与模拟中该残基在结构中的最大口袋体积之间的差值。对于 enspara 中实现的 LIGSITE 计算,我们使用的最小秩为 7,网格间距为 0.7 Å,探针半径为 1.4 Å,最小簇大小为 3 个网格点。为了给残基分配口袋体积,我们计算了每个残基 5 Å 范围内有多少个 LIGSITE 口袋网格点。如果在 MD 模拟中的某个时刻(我们的轨迹以 20 ps 的速率保存,并且我们在每一帧上执行计算)某个残基相对于其在起始结构中分配的口袋体积增加40埃,则我们将其标记为正例。
模型评估
我们使用 5 交折叉验证评估了 GVP-GNN 和 3D-CNN 在基于起始结构预测模拟口袋体积变化方面的性能。我们根据 CAT 代码生成了 5 个折,并限制了折叠之间的重叠,这样除了非常常见的 3.40.50 代码(在每个折叠中出现 1-3 次)外,在拓扑层面上就没有重叠了。我们进行了 5 次独立的模型评估,其中 3 次用于训练,1 次用于验证(即在训练历元之间选择模型并决定选择哪个超参数),1 次用于测试。对于每个分折,都会根据模型在验证集上的 PR-AUCs 选择不同的模型,以便在保留的测试集上进行最终测试。
GVP-GNN 训练
为了优化 GVP-GNN 的性能,我们首先评估了不同的 GVP 网络超参数(即dropout和隐藏层数量)和学习率如何影响单个验证折的性能。这些结果在补充表 2 中提供。我们发现,根据 PR-AUC 评估,低学习率与性能提高相关。然而,其他超参数在感兴趣的范围内对 GVP-GNN 的性能没有实质性影响。因此,我们使用了原始 GVP 论文中报告的相同网络超参数。
为了选择最佳的类平衡方案和批量大小,我们分别计算了 5 个数据集拆分中每个指定验证集的 PR-AUC。为了解决正负残基之间的类不平衡问题,我们训练了模型,包括正类的过采样、负类的欠采样、损失加权以及通过欠采样和过采样绘制平衡的 160 个残基批次。我们还训练了具有中间值的样本和不具有中间值的样本(即,口袋体积差异介于 10 之间的残基A˚3和 40A˚3).为了评估模型性能与批次大小之间的关系,我们使用蛋白质大小的批次、32 个残基大小的批次和 4 个残基大小的批次进行训练。我们发现,当我们使用较小的批量时,模型性能通常更好,但不受类平衡方案选择或包含中间示例的显着影响(补充表 3)。对于每次拆分,我们选择了在验证集上表现最好的GVP-GNN模型(补充表3),并报告其测试集性能(补充表1)。
所有 GVP-GNN 模型均在 AMD Radeon Vega 20 GPU 节点上训练。
3D CNN训练
(略)
一个新的隐秘口袋数据集的构建
(略)
鉴定不太可能形成隐秘袋的残基
蛋白质与配体广泛结晶
高度稳定和/或高度刚性的蛋白质
模拟
进行分子动力学模拟是为了寻找尚未通过实验鉴定的其他神秘口袋,并确保阴性标签的高度可信度。我们对 9 种超刚性蛋白质和 10 种具有广泛全息晶体结构的蛋白质样本进行了模拟。模拟中形成的与袋相邻的候选阴性残基被消除。具体来说,我们消除了指定 LIGSITE 口袋体积大于 20 A˚3 的任何残基。该阈值是通过从 CryptoSite 数据集中查找 13 个holo结构中的残留水平 LIGSITE 口袋体积并选择分布的 第10 百分位数处的值来确定的。剩余的负残基(a)在广泛的全息晶体结构中不结合任何配体,并且(b)在模拟中不与隐秘袋相邻,被用作验证和测试集中的负标签。
PocketMiner 训练和评估(任务 2)
PocketMiner 使用模拟数据进行训练,以预测实验结构中配体结合的神秘口袋(任务 2)。
数据特征化
与之前的预测任务一样,我们使用 MD 模拟中的蛋白质结构进行训练,每个残基都分配一个标签,具体取决于在接下来的 40 ns 模拟中该残基是否形成口袋。由于有多种方法可以计算口袋并将其分配给附近的残基,因此我们使用多种标记方案进行训练。我们使用具有上述超参数的 LIGSITE pocket 检测算法以及具有默认参数 35 的 fpocket 检测工具。我们以两种不同的方式将 LIGSITE 体积分配给附近的残基:(1)通过分配每个残基 5 Å 范围内的所有口袋网格点和(2)将每个口袋网格点分配给最近的残基。
接下来,我们找到了在 40 ns 的模拟过程中残基级口袋体积或药物可达性评分的最大增加(对于 fpocket 成药性评分,我们还使用基于模拟中最大药物评分而不是最大增加的标签进行训练)。为了找到 40 ns 窗口内的最大增加,我们确定了每个结构的口袋体积,相隔 20 ps,以及每个结构的 fpocket 成药性评分,相隔 100 ps。最后,我们根据对已知隐匿位点的口袋形成分析确定的不同阈值对分配的 LIGSITE 口袋体积进行了二值化处理(例如,第一个分配程序的阈值为 20、30 和 40)。由于 fpocket 药物成药性评分已经在 0 到 1 之间,因此不需要进行二值化处理。
GVP-GNN 训练
我们使用与标题为“GVP-GNN 和 3D-CNN 模型训练和评估”的方法部分中所述相同的超参数在 AMD Radeon Vega 20 节点上进行训练。使用不同残留标签(不同二值化阈值下的 LIGSITE 口袋体积变化与 fpocket 成药性分数)、批次大小(1 个蛋白质批次与 4 个残留批次)和类平衡方案(无平衡与 640 的恒定大小平衡抽取)训练的模型使用一组验证实验结构进行比较,包括超刚性蛋白质、具有广泛配体结合晶体结构的蛋白质以及隐秘袋的示例。我们发现,在我们训练的 GVP-GNN 中,使用 LIGSITE 衍生标签训练 20 个 epoch 并使用 fpocket 成药性评分衍生标签细化 1 个 epoch 的模型具有最佳验证 AUC。这个模型被称为 PocketMiner,使用 4 个残基批次进行训练,其中标签是基于在训练的第一阶段将每个口袋网格点映射到其最近的残基来找到分配的 LIGSITE 口袋体积的差异。使用阈值 20 A˚3 对残基标签进行二值化。在训练的第二阶段,使用基于 3 A˚ 截止值分配给每个残基的最大 fpocket 成药性分数来重新标记相同的残基。
模型评估
选择单个模型后,我们使用保留的测试集评估了 PocketMiner 的性能。该测试集包括 24 个具有配体结合隐性口袋的 apo 结构、4 种超刚性蛋白和 7 种蛋白质,它们是广泛配体筛选的对象(参见标题为“从广泛结晶中识别非隐性口袋形成残基”的方法部分)蛋白质和超刚性蛋白质”)。我们的测试集中没有一个蛋白质结构与我们的训练蛋白质具有超过 60% 的序列相似性,但具有 >40% 序列同一性的蛋白质的结构同源性非常差(补充图 7,补充表 5)。